Doença de Fabry
A Doença de Fabry é um distúrbio de armazenamento lisossômico hereditário, ligado ao cromossomo X, causado por uma mutação no gene GLA. Como resultado dessa mutação, há deficiência da enzima alfa-galactosidase A, responsável por decompor determinadas substâncias gordurosas. Essa deficiência leva ao acúmulo de globotriaosilceramida em diversos órgãos.
A Doença de Fabry pertence ao grupo das mucopolissacaridoses ultra raras. A condição pode causar episódios graves de dor ardente e intensa nas extremidades distais, irradiando para outras partes do corpo.
Os sintomas característicos também incluem alterações na termorregulação e na sudorese, mesmo em situações de estresse. Os pacientes geralmente apresentam danos cardíacos ou cerebrais prematuros antes dos 50 anos. Devido à natureza inespecífica dos sintomas, o diagnóstico adequado pode levar vários anos.
Causas da Doença de Fabry
A Doença de Fabry é classificada como um distúrbio lisossômico raro. É causada por uma mutação no gene GLA, que leva à redução significativa ou ausência total da atividade da enzima alfa-galactosidase A.
Logo, a globotriaosilceramida (GL-3) se acumula nas células de vários tecidos e órgãos, já que a deficiência enzimática impede sua degradação. Esse acúmulo compromete o funcionamento dos órgãos afetados e gera complicações.
Essa doença rara é ligada ao cromossomo X, o que significa que ocorre com mais frequência em homens. No entanto, mulheres heterozigóticas também podem apresentar sintomas, com diferentes graus de gravidade. Nelas, as manifestações clínicas geralmente surgem alguns anos mais tarde do que nos homens.
Diagnóstico da Doença de Fabry
Para confirmar o diagnóstico em homens, mede-se a atividade da enzima alfa-galactosidase A no plasma, leucócitos ou fibroblastos. Se for detectada deficiência enzimática, realiza-se o teste genético. No caso das mulheres, como a atividade da enzima pode estar dentro dos níveis normais, o diagnóstico requer análise genética para identificar portadoras do gene defeituoso causador da doença.
Sintomas da Doença de Fabry
Pode se manifestar em dois fenótipos:
- Fenótipo clássico: Mais comum em homens, caracterizado por ausência ou atividade enzimática mínima, com sintomas que aparecem na infância ou adolescência.
- Fenótipo não clássico: Os sintomas surgem mais tardiamente, devido a uma deficiência menos grave da alfa-galactosidase A.
Sintomas que aparecem na adolescência:
- Intolerância a temperaturas altas
- Sudorese diminuída ou ausente
- Hipertermia
- Parestesias (dor ardente intensa nas extremidades)
- Dor abdominal, constipação, diarréia, náuseas e vômitos
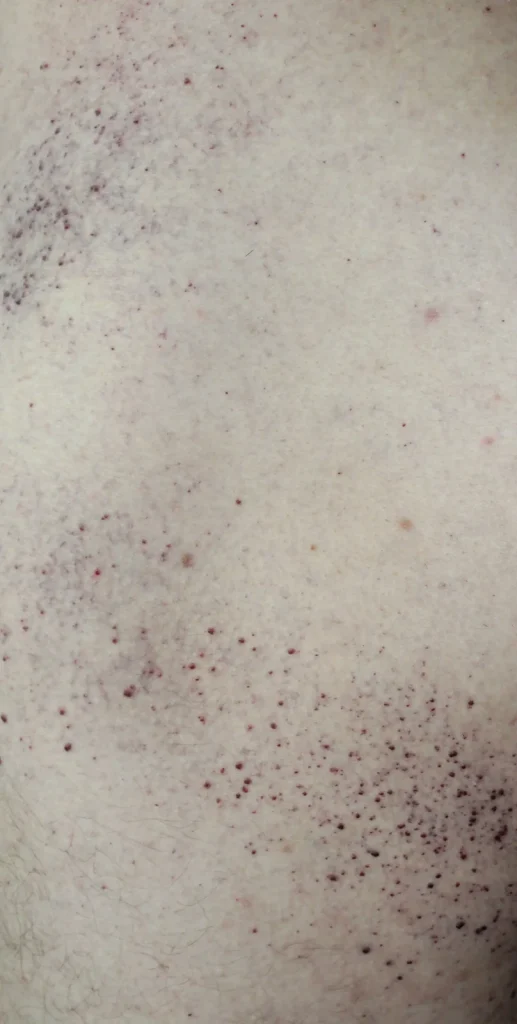
- Lesões cutâneas nas coxas, nádegas e parte inferior do abdômen, na forma de manchas vermelho-arroxeadas (angioceratomas)
- Opacidade da córnea, catarata e perda auditiva progressiva
Sintomas na vida adulta:
- Alterações estruturais e funcionais do sistema cardiovascular (hipertrofia ventricular esquerda, arritmias e distúrbios de condução)
- Comprometimento renal, inicialmente com proteinúria e microalbuminúria
- Fadiga crônica, zumbido e tonturas
Alterações comportamentais como ansiedade, isolamento social, depressão e sentimentos de culpa também são comuns. Muitos desses sintomas aparecem antes do diagnóstico, pois os pacientes com Fabry, como ocorre em outras doenças raras, enfrentam longos atrasos diagnósticos, o que afeta seu bem-estar físico e psicológico. Estudos mostram que a qualidade de vida de pessoas com Fabry é significativamente inferior à da população em geral.
Prognóstico
As causas mais frequentes de morte em pacientes com Fabry são acidente vascular cerebral (AVC) e infarto do miocárdio. Comparados à população geral, os homens com a doença têm expectativa de vida reduzida em 15 a 20 anos, enquanto as mulheres vivem, em média, de 6 a 10 anos a menos.
Opções de Tratamento
O tratamento padrão da Doença de Fabry é a terapia de reposição enzimática (TRE) com agalsidase alfa ou beta. Devido ao envolvimento multissistêmico da doença, terapias de suporte também são essenciais para controlar complicações e prevenir o agravamento do quadro clínico.
A TRE pode desacelerar a progressão da doença e permitir que os pacientes mantenham suas atividades familiares, profissionais e sociais.
Além disso, o migalastate foi aprovado como tratamento para a doença de Fabry. Diferentemente da TRE, que é administrada por via intravenosa, o migalastate é um medicamento oral. No entanto, apenas pacientes com mutações específicas que respondem ao fármaco podem recebê-lo — cerca de 35 a 50% dos pacientes com Fabry.
Terapias de suporte para a Doença de Fabry:
- Controle da dor, principalmente com medicamentos
- Medidas neuroprotetoras (inibidores da enzima conversora de angiotensina e bloqueadores dos receptores da angiotensina)
- Medicamentos antiarrítmicos
- Implante de marcapasso e cardiodesfibrilador (CDI) em casos cardiovasculares graves
- Diálise ou transplante renal em casos de insuficiência renal avançada
O tratamento da Doença de Fabry é contínuo por toda a vida, e a intervenção precoce aumenta as chances de retardar ou evitar complicações graves.
Prevalência
Até 2018, cerca de 73 pessoas haviam sido diagnosticadas com a Doença de Fabry na Polônia. Mundialmente, a prevalência é estimada entre 1 a cada 40.000 a 170.000 indivíduos. No entanto, devido à complexidade dos sintomas e às dificuldades no diagnóstico diferencial, é provável que esses números estejam subestimados.